
Artur Plawgo
Shares of Taysha Gene Therapies (NASDAQ:TSHA) have fallen by 85% since the September 2020 IPO was priced at $20. Performance is an even more brutal -88% since my initial article was published in April 2021 (talk about poor timing).
The company surely suffered during these past couple years, a product of both the overall macro picture as well as biotech bear market and management missteps. Unfortunately, this was a good example of a leadership team that bit off more than it could chew (failing to adequately take heed of cash burn and resources as they sought to build out a 187,000 square foot cGMP manufacturing facility and progress a broad portfolio of CNS gene therapy programs).
To be fair, in the March quarterly report, Founder and CEO RA Session II took the right steps when he announced a workforce reduction of 35% along with narrowing of focus to both GAN and Rett syndrome programs. Research and development expenses tripling to $37.9M was clearly not sustainable in light of the depressed state of the sector as well as the company’s dwindling cash resources.
While highly aware of the significant risks (especially safety) involved in the gene therapy field, after the Astellas collaboration news I chose to reenter the stock ahead of 1H 23 Rett data updates.
Chart
FinViz
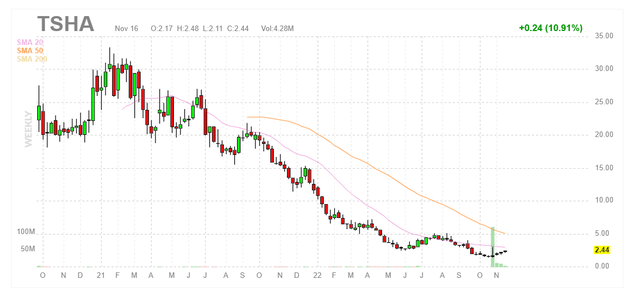
Figure 1: TSHA daily chart (Source: Finviz)
When looking at charts, clarity often comes from taking a look at distinct time frames in order to determine important technical levels and get a feel for what’s going on. In the daily chart above, we can see shares steadily fall from the $10 level to a low of $1 as the company struggled to stay afloat (even proposing a share offering in September only to decide against it, perhaps due to lack of demand or in light of deal discussions with Astellas). Share price briefly jumped to the $3 level on news of the Astellas partnership, only to pull back closer to $2 on news of a $30M secondary offering to follow. My current take is that risk-tolerant investors interested in this name should accumulate dips in the near term (below the $3 level).
Overview
Founded in 2020 with headquarters in Texas (115 employees), Taysha Gene Therapies currently sports enterprise value of ~$50M and estimated Q3 cash position of ~$110M (if current secondary offering is successful) providing them operational runway into 1H 2024. Per filing prospectus, there were ~41M shares outstanding as of June 30 (excluding 7.2M shares from recent Astellas private placement and shares to be issued in current secondary offering).
Taysha is utilizing a specific validated gene therapy technology (AAV9) that’s proven its efficacy, safety and durability in multiple diseases via peer data. AAV9 is coupled with novel targeted payload design and intended to address a range of monogenic CNS disorders. Again, after cutting costs, the company is mainly focusing on just the GAN and Rett syndrome programs. They do intend to conduct small proof-of-concept studies in CLN1 disease and SLC13A5 deficiency. Another benefit is highly scalable manufacturing (keep in mind they paused or cancelled construction of a new 187,000 square foot cGMP facility in North Carolina as part of cost cuts).
Founding collaboration is with UT Southwestern Gene Therapy program, as Dr. Steven Gray headed the first intrathecal dosed gene therapy candidate. This collaboration gives Taysha access to scale and expertise across their gene therapy portfolio. Intrathecal delivery (injection in spinal canal) is the chosen route of delivery as doctors have given intrathecal medicine for decades in outpatient setting across multiple modalities – it allows them to target the CNS broadly and avoid neutralizing antibodies by starting on the right side of the blood brain barrier. Management refers to multiple studies run by the likes of AveXis in SMA, Amicus, and the list goes on regarding validation of this approach and technology. Taysha utilizes highly scalable HEK293 manufacturing process with excellent yield (provides right balance between scalability, yield and risk, safe and effective across multiple serotypes in clinic to date, feel strongly does not come with inherent toxicities that come with other systems).
Lead GAN program originated from Chief Scientific Officer Steven Gray’s lab and is the first intrathecally dosed gene therapy study in history. As for opportunity in GAN, there are around 2400 prevalent patients in US + EU ($500M to $1B potential peak sales). GAN is an ideal target for the company’s approach because it is a small gene that fits well into AAV9 capsid, has high transduction at target organ and there’s evidence that low level expression of gigaxonin may restore function in humans. This construct is clearly a model for other candidates in pipeline to treat additional CNS diseases. A rare inherited genetic disorder, GAN patients are typically symptomatic before the age of 5.
Corporate Slides
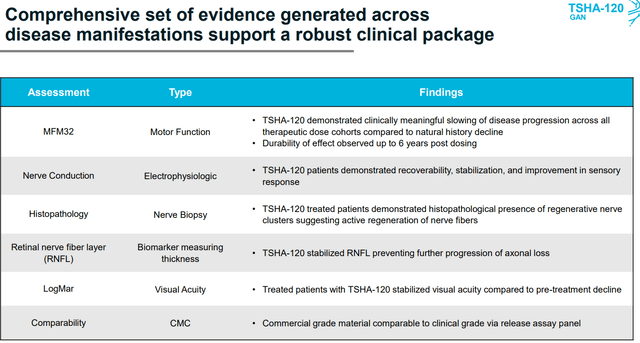
Figure 2: TSHA-120 GAN program summary (Source: Updated slides)
As for primary clinical measure, the MFM32 is a validated quantitative scale (motor function measure with four-point change considered to be clinically meaningful including for other indications like DMD, SMA, cerebral palsy, etc). In this 32-item scale, a higher score is better, and for frame of reference, natural history study for GAN shows an 8-point decline annually (in 45 patients, age 3 to 21 years old). MFM32 will be the primary endpoint for a confirmatory trial. Safety and efficacy results here are promising, especially two highest doses which showed meaningful slowing of disease progression along with dose dependent effect (statistically significant). Highest dose of TSHA-120 resulted in an 8-point improvement vs. historical control, with 6-point improvement occurring at second-highest dose (keep in mind 4-point change is considered meaningful). TSHA-120 was considered well tolerated and six patients have been followed for more than three years (seeing sustained dose dependent improvements in MFM32 scores). Further analysis shows nearly 100% probability of clinically meaningful slowing of disease progression compared to natural history.
As for TSHA-102 for Rett syndrome, this is one of the most common genetic causes of intellectual disabilities in women. Rett is caused in most cases by loss of function mutations in the MECP2 gene. MECP protein is essential to neuronal and somatic function in the brain. For effective treatment, MECP2 expression needs to be titrated to correct the MECP2 deficiency while avoiding adverse effects associated with having too much MECP2. Regulation of MECP2 to appropriate levels needs to occur on a cell by cell basis, thus a proprietary method of transgene regulation was developed called miRARE (miRNA targeting panel that binds to endogenous downregulatory microRNAs that are activated in presence of high levels of MECP2). Preclinical studies for TSHA-102 showed favorable tolerability and increased survival in knockout mouse model. The estimated prevalence of Rett syndrome is 25,000 patients in the US and EU.
Astellas Deal and Pipeline Update
On Oct. 21, the company entered into an Option Agreement with Astellas Gene Therapy, granting them an exclusive option to obtain an exclusive, worldwide, royalty and milestone-bearing right and license to both the GAN and Rett Programs.
GAN option is exercisable anytime from the present through receipt of the formal minutes from Type B end of phase 2 meeting between Taysha and the FDA (formal communication on the outcome of this meeting expected in January 2023).
As for the Rett option, it can be exercised anytime through receipt of clinical data from the female pediatric trial.
Corporate Slides
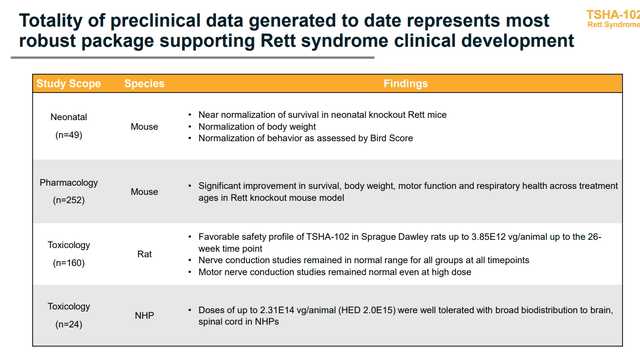
Figure 3: TSHA-102 Rett program summary (Source: Updated slides)
Provisions of this collaboration are quite interesting, as terms of license agreement have not been negotiated yet and this will occur after receipt of FDA minutes or Rett data. During the Rett Option period, Taysha agreed not to solicit offers or proposals from other companies (seems like Astellas wants to lock this up) and terms include a “Change of Control” clause giving Astellas the right of first proposal for any potential buyout or transaction.
In return, Astellas paid them $20M upfront payment and purchased $30M of stock (over 7.2M shares) via private placement.
As for the associated clinical update, Taysha notes that the Type B meeting for GAN program has been scheduled for Dec. 13 with the FDA and so receipt of formal meeting minutes are expected in mid-January.
With respect to TSHA-102 for the treatment of Rett syndrome, originally the company was going to report early phase 1/2 data at the end of this year (just safety). However, after this deal with Astellas, they now expect to report preliminary safety and efficacy clinical data from the first cohort of adult patients 1H 23. They also plan to initiate the female pediatric clinical study 1H 23.
Detailed breakdown of finances states that they had just $34.3M of cash as of Sept. 30 (quite desperate considering Q2 net loss was ~$33M). However, this does not include $50M payment from Astellas as well as the expected $30M of proceeds from secondary offering that followed. Interestingly enough, the filing for the secondary offering shows that insider Paul Manning/PBM Capital has indicated desire to purchase up to $5M of shares in the financing.
From are my notes from the associated call:
- $30M investment was made by Astellas for 15% of outstanding shares (implies $200M valuation). This was confusing in the initial press release, where investors and all news sources cited a $50M investment. However, the deal format entails a $20M payment and $30M investment via private placement.
Astellas receives certain rights to change of control until after Rett data (whether Astellas wants to license the two candidates or outright buyout the company, they are able to counter should another pharma make an offer).
- Regarding Rett syndrome program, which is what most attracted Astellas, toxicology data showed favorable safety at up to 4x higher dose in NHP (non-human primates) than starting dose in humans. Also, there was no evidence of dorsal root ganglia inflammation. NHP data at all doses studied showed the gene therapy candidate was well tolerated with broad biodistribution.
- The effect on Rett knockout mice up to 20 days post birth (50% through their normal life span) was striking. In some cases they allowed mice to mature to a level of adulthood (accumulating significant level of disease burden) before intervening with treatment. Yet, they still saw improvements across multiple parameters in survival, respiratory function and motor function.
- I’m a fan of the phase 1 trial design, as I think it’s unethical for any patient to not receive treatment. Six adult Rett patients are being enrolled and randomized to treatment with TSHA-102 or delayed treatment control (three patients on drug, three patients with delayed treatment). This way, the study will be more robust and allow them to compare treated patient to non-treated.
- Capital injection is meant to allow them get to meaningful data sets in Rett adult AND pediatric patients.
The deal came out of conversations over the past four to five months with Astellas.
If all goes well with GAN regulatory-wise, they could commercialize it in 2024.
Astellas had access to “all available data” that Taysha was in possession of to make this decision (sounds to me like they saw initial human data).
Q3 Update
For Q3, cash position of $34M (+ $50M from Astellas and $25.6M from secondary offering= $109M total) is still insufficient compared to net loss of $26M. However, it’s much better than teetering on the brink of bankruptcy, which is where the company was previously. Research and development expenses were sliced by more than half to $16.4M, while G&A fell to $8.7M. Management is guiding for operational runway into Q1 2024, but I expect another financing by mid 2023 after Rett data update.
$28M secondary offering was priced at $2/share (14M shares), which was very disappointing given the prior runup. I definitely jumped the gun here in terms of pulling the trigger versus even showing a day or two more of patience, but that error/lack of prudence is on me and me alone.
On the bright side, Director of the Board Paul Manning bought ~$3M worth in the offering and now owns nearly 7M shares or 11.2% of the company.
They continue to guide for TSHA-102 data update in first cohort of Rett syndrome adult patients 1H 23 (initiation of female pediatric cohort around the same time frame). This language sounds increasingly confident, as prior pediatric cohort was only going to be started if adult data was good.
Regulatory update for TSHA-120 in GAN is expected mid-January 2023 (devil’s advocate is that despite the strong data, FDA asks for more info on CMC or even more clinical data). We will see.
As for the conference call and Q&A, here are a few nuggets:
- In regard to pivotal trial design for Rett syndrome, it seems too early to ask. However, management states that this would focus on pediatric population as this is the key population of interest (also applicable to wider population). Assessments are similar such as looking at frequency of seizures, how severe and how long they are. Another bucket is respiratory outcomes which they demonstrated improvements very clearly in animal models. They also look at communication ability, behavioral scale, biomarkers, EEG, etc. Buckets are same from adults to children, but specific assessments are a bit different.
- With regard to safety, toxicology studies showed they gave 4-fold dose over the initial starting dose (5e14 total vg) in both NHPs and rats over three- and six-month period (all the way up to 2e15 equivalent). 4-fold dosing of initial clinical starting dose they saw no adverse toxicological findings. Thus, they have absolute confidence in 5e14 level in humans will not result in overexpression of MECP2 (key concern).
- Vast majority of patients enrolled in pediatric study will be over the age of three or four (won’t need to change dose because size of child’s brain has reached size of adult’s). Again, 5e14 starting dose in both.
- As for bar for success in clinical data, safety will be extremely important (construct is designed to make sure patients get the level of MECP2 they need, to see significant improvement across spectrum of disease and doing that safety in the presence of wild-type MECP2). 50% of these patients’ cells are normal, 50% are null or not producing MECP2. Too much MECP2 is toxic, so the construct is designed to express the needed level of MECP2 on a genotypic cell-by-cell basis. Totality of data will be important in terms of efficacy and they’re expecting preliminary efficacy. They expect children to do better than adults because treating developing brain results in better outcomes in animal studies than adult brain. Also, animal studies across many different ages of mice showed it was clear the younger mice did better than the older ones. Autonomic dysfunction (rapid breathing, hypoxic excess go blue because not breathing) is one key area that if moderated by treatment would be clinically meaningful for patients and families. If they can reduce seizure burden or bring modicum of functionality to hand function, these are additional examples of things that are meaningful. This is a global, neurological developmental disease so they are looking at a wide variety of measures.
- Pediatric study will be a global study in multiple countries around the world. Gateway for moving into pediatric study is getting some level of follow up for multiple months in adult study to give them confidence to include in data package for pediatric trial.
- Rett advocacy group puts worldwide prevalence at ~350,000 patients and recruiting for such a study should not be a problem per management. Company calculated 25,000 to 35,000 in US and EU alone.
- Analysts asked what toxic adverse events would look like due to MECP2 overexpression in humans, but management reminds him that they did not see this in animal studies at very high doses.
- Initial adult data would be in three to five patients to give them confidence to move forward into children. Natural history in Rett syndrome is some of the most well-characterized out of any disease. For them, it’s getting to three to six patients including delayed treatment patient.
- For GAN program (the most advanced asset), their intention is to file an MAA in Europe and they believe current data package checks all the boxes regarding FDA guidance for gene therapies in neurodegenerative diseases. However, to my eyes playing devil’s advocate, they still don’t sound overly confident here (perhaps FDA requests more information, more data implying delays, etc).
Final Thoughts
To conclude, listening to the Astellas deal conference call twice (along with referring to my prior notes) convinced me to hop on for a pilot position with a view toward Rett data updates 1H 23 along with progression into the pediatric clinical study. The Rett patient community currently has nothing else out there for them after Novartis’ gene therapy program was discontinued last year (see their reaction) and so I’m (not only as an investor) rooting for results (here or elsewhere) that bring them hope.
For readers who are interested in the story and have done their due diligence, TSHA is a Speculative Buy and I suggest accumulating dips below $3. A conservative strategy could be to purchase half of desired exposure presently, then wait for Type B meeting notes in January for the GAN program before considering further action.
One risk that comes to mind is additional dilution in 2023 (they only have runway to Q4 2023 or 1H 2024 at latest, which also depends on the success of current secondary offering where pricing or demand could be weak in the worst-case scenario). Safety/tolerability issues including patient deaths or Grade 4 adverse events that unfavorably skew risk/benefit of gene therapy treatments has always been a key concern for this area of biotechnology (just look at Audentes Therapeutics’/Astellas’ XLMTM program still on clinical hold). Disappointing data in Rett would weigh very heavily on shares, as despite promising long-term results for GAN I don’t think the market will give much credit to the latter until approval. Regarding GAN Type B meeting with the FDA, it’s clear the company is hoping for a win with green light to proceed for regulatory filing. The FDA lately has been quite amenable to rare diseases where nothing is approved, such as recent green light to Relyvrio’s recent approval in ALS changing the fortunes of Amylyx (AMLX). However, playing devil’s advocate, the FDA could request a pivotal study be run first instead of as a confirmatory trial. Other issues could also arise, such as if they have questions on the material used in clinical study vs. that intended for commercial launch (bridging study). One final risk, as alluded to above, is that current secondary offering fails (insufficient demand) or is priced at unfavorable levels (causing share price to fall further).
")






")







{kind=link}