
Shutter2U/iStock via Getty Images
Intro
Relmada Therapeutics (NASDAQ:RLMD) is advancing its NMDA antagonist, REL-1017 (esmethadone), as a novel antidepressant for the treatment of major depressive disorder (MDD). After reporting strongly positive Phase 2 results in late 2019, the company announced the failure of two Phase 3 trials in late 2022, cratering the stock from $1+ billion down to its current micro cap levels (~$100 million).
Despite the topline failures, we believe the totality of evidence indicates that REL-1017 has strong underlying efficacy, and that the two Phase 3 trial failures can be attributed to abnormally high placebo responses that were caused by a confluence of management’s prioritization of enrollment speed over patient quality, suboptimal trial design, COVID-related increases in situational depression, and a small number of outlier clinical sites that reported extremely high placebo responses. We believe management has taken meaningful steps to remediate these issues, and believe the market is currently underestimating the likelihood of positive results in the ongoing Phase 3 trials, RELIANCE-II and RELIGHT, which are expected to read out in mid-2024 and year-end 2024, respectively.
Investment Thesis
In both of the failed Phase 3 trials, REL-1017 delivered ~15 pt improvements on the gold-standard Montgomergy-Asberg Depression Rating Scale (MADRS), a strong treatment effect that is in-line with the Phase 3 results of many approved antidepressants. However, both trials also reported abnormally high placebo responses of 13-14 pts, significantly higher than the ~8-12 pts considered to be typical of adequately controlled antidepressant trials, causing both studies to fall short of statistical significance.
Relmada has shared a number of post-hoc analyses and details to explain the high placebo responses, including that a majority of patients enrolled were recruited from questionable sources (e.g. social media advertisement, internet searches), and that one of the highest-enrolling trial sites reported placebo responses of 20+ pts in both Phase 3 trials.
Management has taken ownership of the various deficiencies and made specific protocol and operational changes to address them, including requiring medical/pharmacy records to verify MDD diagnosis and excluding lower-quality trial sites altogether. With these improvements, we think REL-1017—which has consistently reported strong 15-17 pt MADRS improvements across Phase 2, Phase 3, and recently-reported open label studies—will be able to separate from a properly-controlled placebo to deliver positive results in its currently ongoing Phase 3 studies, RELIANCE-II (~75% of patients enrolling under amended protocol) and RELIGHT (100%).
As an investment opportunity, Relmada’s setup reminds us of Axsome (AXSM) in late-2021/early-2022 (our May 2021 Axsome article), with both companies being overly punished by investors after a series of setbacks. While Relmada’s primary concern (i.e. delivering statistical significance in its forthcoming Phase 3 trials) is riskier than Axsome’s (manufacturing- and capacity-related setbacks at the FDA), we believe the totality of evidence supports REL-1017’s underlying efficacy and are confident in Relmada’s turnaround story.
At ~$100 million of market cap, and with sufficient cash to get through both ongoing Phase 3 trials, we believe the market is significantly underestimating Relmada’s probability of success and expect stock to creep up as we apprach the Phase 3 readouts in 2024. In the event that both RELIANCE-II and RELIGHT are successful, we would expect Relmada to trade back up to its previous $1+ billion market cap peak, which would correspond to ~$33/share, representing ~10x upside potential in 2025.
REL-1017 and Methadone
REL-1017’s active ingredient is esmethadone, the s-enantiomer of the synthetic opioid methadone. Methadone, originally approved as an analgesic in 1947 and for the treatment of opioid addiction in 1972, refers to the racemic mixture of the s- and r-enantiomers of methadone, named esmethadone and levomethadone, respectively. Enantiomers, also called optical isomers, are mirror images of the same molecule (i.e. “sister molecules”) but are not superimposable and can exert distinct pharmacological effects from one another.
REL-1017/esmethadone is a similar approach to that of esketamine, the active ingredient of Johnson & Johnson’s (JNJ) Spravato, which is the s-enantiomer of racemic ketamine, which was first approved as an anesthetic in 1970. However, unlike the two enantiomers of ketamine—which appear so far to have relatively subtle pharmacological differences—esmethadone has proven to have distinct phramacological effects versus levomethadone.
REL-1017 was demonstrated to have at least ~20-fold lower affinity for the mu opioid receptor (associated with analgesia, pleasure, and addiction) than levomethadone (Codd, et al., 1995). Thus, REL-1017 has been termed the opioid-inactive enantiomer of methadone, though this label may be technically inaccurate as it still retains some activity at mu opioid receptors, though thought to be insignificant (Fava, et al., 2023).
Most importantly, research has shown REL-1017 does not cause reinforcement, physical dependence, or withdrawal symptoms indicative of addiction potential in animal studies (Henningfield, et al. 2022), in contrast to racemic methadone (Vajda, et al. 1975; Ling, et al., 1984).
Relmada has also conducted human abuse potential (HAP) studies comparing esmethadone at up to 150 mg (6x the Phase 3 dose) to 40 mg of oxycodone in recreational oxycodone users, 0.5 mg/kg IV of ketamine in recreational ketamine users, and 300 mg of dextromethrophan (DXM; the active ingredient in the recently approved antidepressant Auvelity, though at ~1/6th of the dose). REL-1017 was found to be statistically equivalent to placebo on a “drug-liking” visual analog scale (VAS), in contrast to oxycodone and ketamine (Shram, et al, 2023). REL-1017’s drug-liking was also found to be statistically significantly lower than DXM.
Company Poster Presentation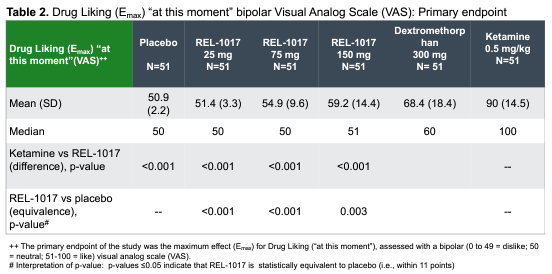
Relmada and the scientific community’s work on REL-1017 lead the FDA to state, “the d-isomer (esmethadone) lacks significant respiratory depressant action and abuse liability”, confirming that REL-1017 is functionally opioid-inactive and is viewed as such by regulatory agencies.
Mechanism and NMDA Receptor
Relmada states that REL-1017’s antidepressant actions are mediated primarily by its uncompetitive antagonism of the NMDA receptor, the same (putative) primary mechanism of action of ketamine and DXM. NMDA antagonism has become one of the “hot” mechanisms in neuropsychiatry in recent years, leading biopharma companies to emphasize its central role in their drug’s antidepressant effects, when the reality is less clear.
For example, multiple studies have found Spravato/esketamine’s antidepressant effects to be dependent on mu opioid receptor activation (Heifets, et al., 2021), and Auvelity’s components (DXM and bupropion (i.e. Wellbutrin)) have serotonin reuptake inhibition, dopaminergic, and noradrenergic effects in addition to NMDA antagonism.
Similarly, REL-1017’s antidepressant effects may not be exclusively attributable to its NMDA antagonism. REL-1017 is an NMDA antagonist, but also appears to have appreciable activity on calcium channels, SERT, sigma receptors, and mu opioid receptors.
REL-1017 was found to increase circulating brain-derived neurotrophic factor (BDNF) levels by between 2x and 17x after 10 days of treatment in a Phase 1 healthy volunteer study (De Martin, et al., 2021). Increased BDNF expression is considered to be a consequence of NMDA antagonism (seen with ketamine, memantine, etc.) and has been demonstrated as a primary antidepressant mechanism of ketamine. Similarly, REL-1017’s antidepressant effects have been found to be dependent on the upregulation of mTORC (mammalian target of rapamycin complex) and synaptic protein expression in the medial prefrontal cortex, a downstream effect of BDNF upregulation (Fogaca, et al., 2019).
Phase 2 Results Presentation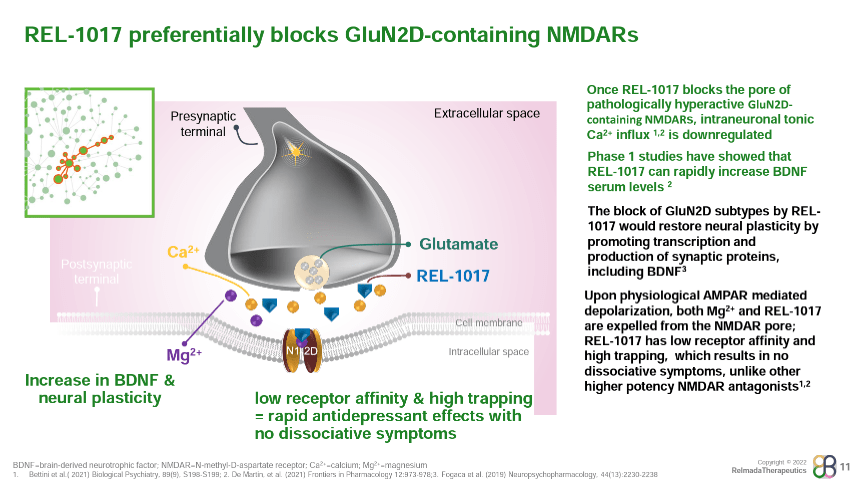
Where REL-1017 may differentiate among NMDA receptors is in its binding profile. REL-1017 has relatively low affinity for NMDA receptors compared to other NMDA antagonists (shown below), but has been shown to preferentially inhibit tonic signaling of the GluN2D NMDA receptor subtype, which is hypothesized to be a key underlying mechanism of glutamatergic dysfunction in psychiatric disorders (Bettini, et al., 2021).
Poster Presentation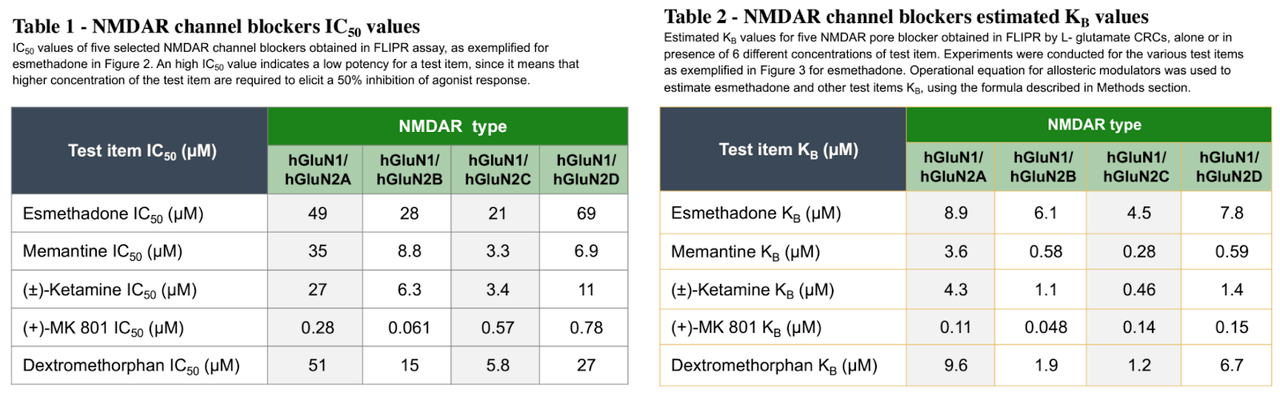
REL-1017 has also been found to have high “trapping” at NMDA receptors, a measure of how slowly a molecule exits its target receptor. The combination of low affinity and high trapping is relatively unique among NMDA antagonists. Ketamine, for example, exhibits both high affinity and high trapping, while memantine exhibits high affinity with low trapping.
Relmada and researchers hypothesize that REL-1017’s combination of low affinity and high trapping could explain its ability to retain antidepressant effects without causing the dissociation and psychotomimetic effects seen with stronger NMDA antagonists like Spravato/ketamine and MK-801 (investigated by Merck in schizophrenia and depression but found to have too many serious side effects).
With its insignificant activity at opioid receptors, lack of abuse potential, benign safety profile, and lack of psychotomimetic/dissociative effects, REL-1017 presents a promising clinical profile in MDD.
Phase 2
REL-1017’s Phase 2 tested 62 total patients randomized 1:1:1 to placebo, 25 mg, and 50mg. The study was conducted on inpatients (patients hospitalized with severe depression), with dosing for seven days and observation out to Day 14. The data was reported in October of 2019, with REL-1017 delivering robust antidepressant effects as early as Day 4, and outperforming placebo by an impressive 9.4 and 10.4 pts at Day 14 in the 25 mg and 50 mg groups, respectively.
Corporate Presentation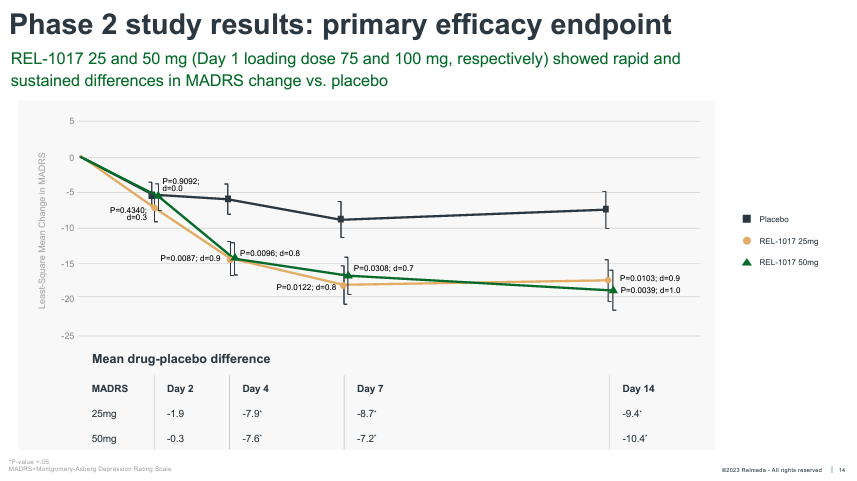
These effects are robust both in terms of absolute reduction of depression symptoms, with absolute MADRS reductions exceeding ~17 pts for both doses, and on a placebo-adjusted basis, especially considering patients were dosed for only seven days.
The average placebo-adjusted MADRS reduction of approved antidepressants is considered to be ~2-3 pts, versus ~10 pts for REL-1017 across both Phase 2 doses. These data also delivered a Cohen’s D effect size of 0.9-1.0 for the two doses, far stronger than the 0.3 average effect size of antidepressant therapies (Cipriani, et al., 2018).
REL-1017 onset of action was also much more rapid than traditional antidepressants, reaching statistical significance as early as Day 4. REL-1017 also reported an impressive proportion of clinical responses (50%+ reduction from baseline on MADRS) and remissions (MADRS < 10) at Day 14.
Corporate Presentation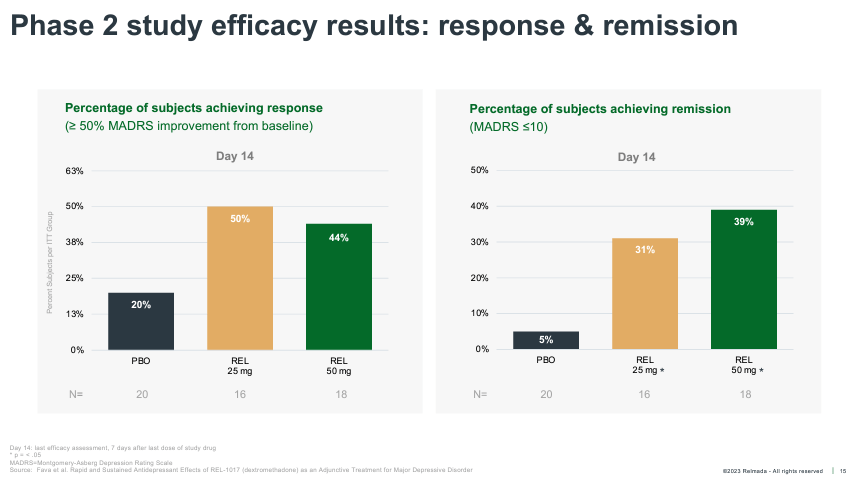
No psychotomimetic or dissociative effects or serious adverse events were reported in the trial.
While the results of the Phase 2 were highly impressive, in hindsight, there were a number of reasons to expect the results to temper going into a larger Phase 3, including the small sample size of only 62 patients and an inpatient population which is known to respond more dramatically to intervention.
Still, the Phase 2 data prompted a more than 500% increase in Relmada’s stock price over the course of a couple months, taking its market cap above $600 million (+566% vs. current price). The company also took the opportunity to raise $115 million (at $30/share), and selected 25 mg as the sole Phase 3 dose of REL-1017.
Phase 3 and RELIANCE-III
Relmada announced the initiation of its first Phase 3 trial, RELIANCE-I, in December 2020, followed by RELIANCE-II in April 2021. Both studies dosed REL-1017 as an adjunct therapy in MDD, meaning as an additive therapy in patients with inadequate responses to existing antidepressant treatment. Both studies were designed to enroll 364 patients each (1:1 placebo vs 25 mg) and had a primary endpoint of MADRS reduction at Day 28. The company also initiated a Phase 3 monotherapy trial, RELIANCE-III, in October 2021.
Relmada’s market cap climbed from ~$600 million to over $1 billion of market cap by the 2H22 in anticipation of the Phase 3 readouts following the impressive Phase 2 results.
Interestingly, the monotherapy RELIANCE-III trial was the first of the Phase 3 trials to report its topline results in 232 MDD patients in October 2022 (despite being the last of the three trials to initiate). The company reported that RELIANCE-III failed to meet its primary endpoint, with REL-1017 delivering a 14.8 pt MADRS reduction at Day 28, compared to a 13.9 pt reduction for placebo, a numerical but not statistically significant improvement.
In the press release announcing the results, Relmada stated that “paradoxical results were observed in certain study sites, where placebo dramatically outperformed REL-1017”. The press release also included a post-hoc, exploratory analysis which excluded the clinical sites that reported implausibly high or low placebo responses, defined as less than 3 pts or greater than 14 pts improvement on MADRS. In this analysis, REL-1017 outperformed placebo by 4.9 pts on MADRS (p<0.05), a strongly statistically significant result. These results are relevant in our opinion given that a placebo response of 14+ pts would be considered firmly “uncontrolled” versus the 8-12 pts typical of antidepressant trials.
A month later, on the 3Q22 earnings call, CEO Dr. Sergio Traversa shared that the top enrolling site in RELIANCE-III had a mean placebo response of 23 pts, effectively double the expected placebo response in antidepressant trials. For context, the most drastic example we could find of a high placebo response was Sage Therapeutics’ WATERFALL trial of zuranolone, which reported a 15.1 pt improvement for the placebo group at Day 15, a similar result to those seen in multiple of Sage’s zuranolone trials, likely on account of the unorthodox nature of zuranolone and its dosing.
The failed topline data caused Relmada’s stock to decline ~80% from ~$30/share to ~$6/share, which then continued to fall towards $3/share (in line with current levels) after CEO Traversa stated on the 3Q22 earnings call that the high-enrolling clinical sites from RELIANCE-III also recruited significantly in RELIANCE-I.
RELIANCE-I
As foreshadowed, RELIANCE-I, which reported topline data in December 2022, also failed to meet its primary endpoint. The trial was halted early by the data monitoring committee (for futility) and thus enrolled only 227 patients. The REL-1017 treatment group saw a 15.1 pt reduction on MADRS at Day 28 versus 12.9 for placebo, a larger numerical improvement than RELIANCE-III, but still falling short of statistical significance.
Corporate Presentation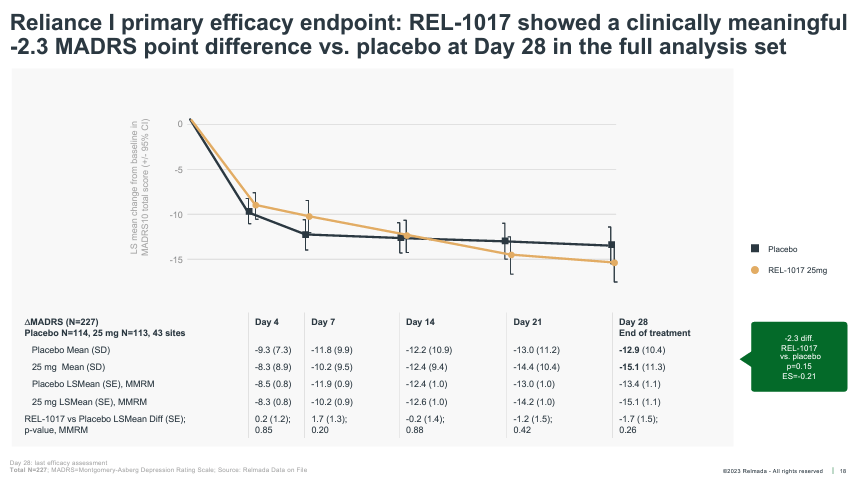
The company stated in the press release that the same “limited number of high enrolling sites with implausible placebo response (from RELIANCE-III), also affected RELIANCE-I”, leading to failure to reach statistical significance. While it sounds like a boilerplate explanation following a failed trial, the data and post-hoc analyses presented in the following weeks and months, in our opinion, substantiate Relmada’s claim.
What we believe to be the most compelling of the post-hoc analyses was contained in the same PR and discussed on subsequent earnings calls:
December 2022 Press Release
This post-hoc analysis excluded only the 42 patients (n=185 vs. n=227) that were enrolled at the two problematic high-enrolling sites, and yet the response data, especially for the REL-1017 treatment group, improved dramatically. The 16.7 pt improvement for the REL-1017 treatment arm is essentially in-line with the effects size seen in the Phase 2 data (~17 pts), and was strongly statistically significant (p=0.019).
While the exclusion of these two sites normalized the placebo group slightly (from 12.9 to 12.6 pts), 12.6 pts is still an abnormally high placebo response that indicates RELIANCE-I was fundamentally flawed on an enrollment, protocol, and/or execution level (discussed further in the next section).
Interestingly, despite the adverse circumstances, the whole-study data (no post-hoc analysis) still managed to reach statistical significance, albeit narrowly, on the key secondary endpoint of clinical response rate (50%+ MADRS reduction) at Day 28, while missing on clinical remission (shown below).
Corporate Presentation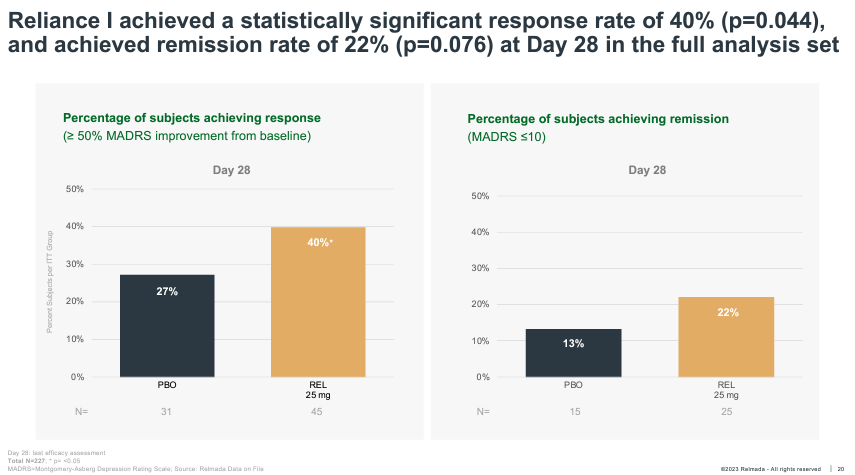
Additionally, in the following months, Relmada shared the pre-specified “per protocol” data set, which excluded only patients that either did not complete the full 28-day study or had other major study protocol deviations. Shown below, the per protocol data set excluded only 29 patients and showed a widening of the placebo-adjusted response to 3.1 pts, which was nearly statistically significant (p=0.051).
Corporate Presentation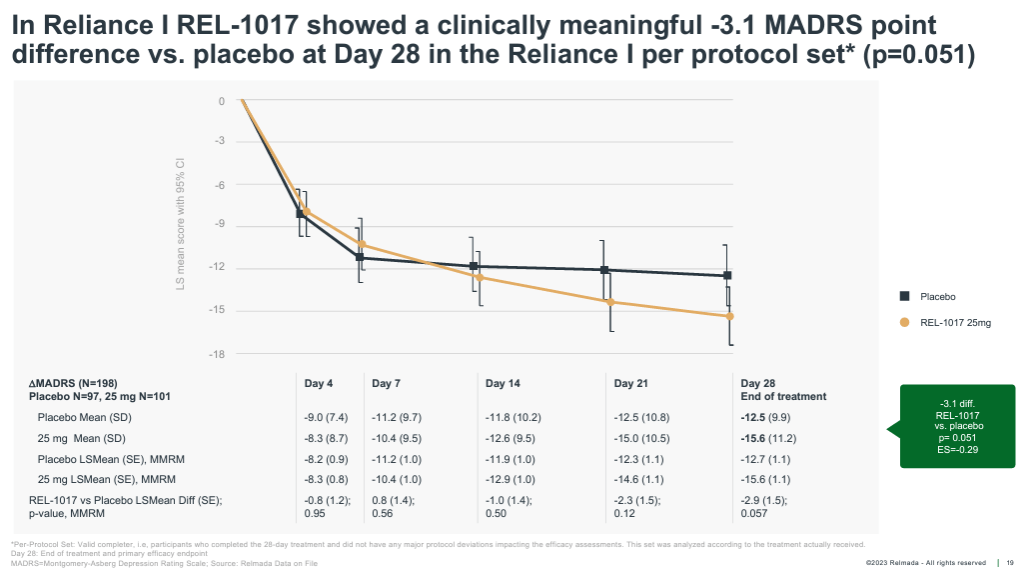
Lastly, Relmada performed a post-hoc analysis that broke patients into those enrolled from “verifiable” vs. “unverifiable” sources. Verifiably-sourced patients included those that were past or present patients at the clinical site, in the sites’ database, or referred from a healthcare professional (HCP). Unverifiably-sourced patients were defined as those sourced from internet searches, social media, TV/radio ads, recruitment companies, and referrals from a family member, friend, or other trial patient.
There ended up being 86 verifiable and 130 unverifiable patients (which speaks to Relmada’s prioritization of enrollment speed over quality in RELIANCE-I). While this analysis is inherently more discretionary than those discussed above, the results (shown below) are interesting, again achieving strong statistical significance (p=0.016). In the unverifiably-sourced patients, the placebo paradoxically outperformed the REL-1017 group by more than 1 pt at Day 28, showing an implausibly high 14.3 mean MADRS improvement.
Corporate Presentation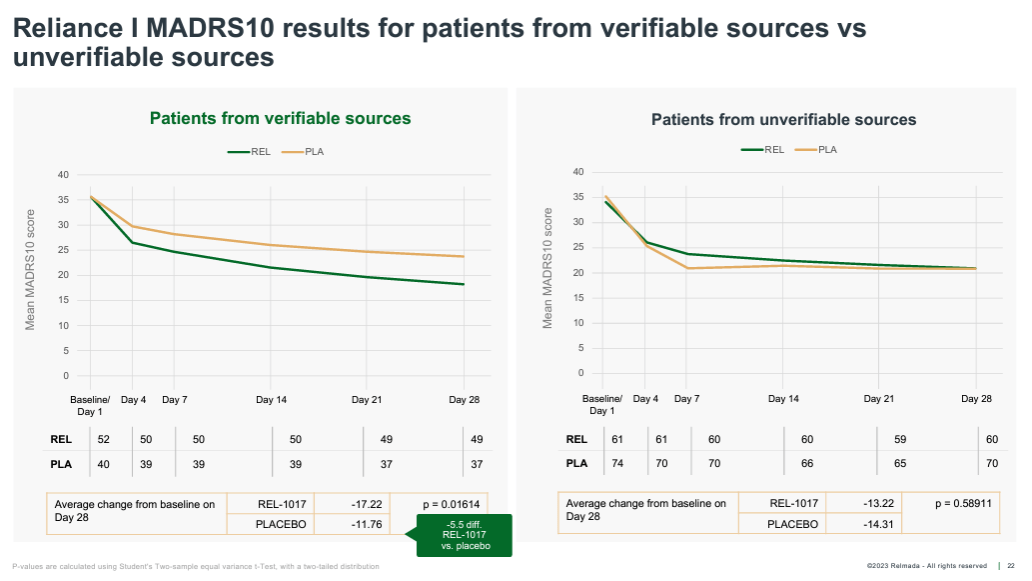
In our view, the various data and sub-analyses provide compelling evidence that the failures of RELIANCE-I/III were driven primarily by management’s inattention to enrollment quality and proper trial oversight, rather than a lack of REL-1017’s underlying efficacy.
Regrouping
Relmada management has been straightforward and transparent in their efforts to identify and correct the deficiencies of RELIANCE-I/III. To this end, the company added Dr. Cedric O’Gorman as CMO in January 2023, a former Axsome Therapeutics, Intra-Cellular Therapeutics, and Roche/Genentech executive with extensive experience in clinical neuropsych research.
We believe Dr. O’Gorman has brought clarity and confidence as point man on the issue of clinical quality control. At the Goldman Sachs Healthcare conference in June 2023, Dr. O’Gorman stated that he believed there were three primary reasons for the abnormally high placebo responses: the enrollment sites, the study protocol, and the subjects. Dr. O’Gorman went on to explain:
“Well, when you work in MDD for a long time, decades, right, in my case, it’s maybe 2 decades now, you become very familiar with sites, site networks that have a number of different sites within their network, which is becoming much more and more common. And you become very familiar with the PIs (primary investigators) by name. And so like in any industry or in any career, you know who have reputations as being better performers, more conscientious, more professional, focused on true patient type as opposed to those, which it’s a business after all, and we can understand motivations. But you want to be very careful about those that have kind of a reputation to be a research mill. And they’ll throw any amount of money on advertising just to get patients into the study.”
On the RELIANCE-I results call, Dr. Maurizio Fava, Chief of Psychiatry at Mass General and principal investigator for RELIANCE-I, shared that the two problematic clinical sites reported placebo responses that were ~10+ pts better than the REL-1017 group in both RELIANCE-I and RELIANCE-III. It was also revealed that one of the two sites reported a 20+ pt improvement for the placebo group in both RELIANCE-I and -III.
Fava stated: “As a clinical investigator, I have some concerns about the ratings, the ratings conducted at those two sites. That would be my guess that something happened in the ratings that would explain why in the two parallel studies, the same problem, the same implausible pattern, was reported.”.
The fact that the same site reported 20+ pt placebo improvements for both monotherapy and adjunctive (which should include less situationally-depressed or falsely-diagnosed MDD patients) raises serious questions about the integrity of the site.
Though it is difficult to speculate exactly why the data reported from these two clinical sites were so highly unusual, management clearly believes that these two sites were compromised in some way. The fact that excluding these two sites from the RELIANCE-I data results in a strongly statistically significant result speaks to their influence on the study.
Study Design and Patient Enrollment
Beyond the two most problematic sites, the ~13-14 pt placebo responses seen in RELIANCE-I/III indicate a study that was suboptimally designed, enrolled, and executed on a fundamental level.
On the 3Q22 earnings call, Dr. Traversa stated that “pretty much all” of the patients enrolled at the two high-enrolling sites were recruited via social media advertising, which leads us to believe that the majority of the patients enrolled across all RELIANCE-I/III trial sites were enrolled via social media and similar sources.
As seen in the “verifiable” vs. “unverifiable” post-hoc analysis, ~60% of patients were enrolled from questionable sources, including social media/TV/radio advertisements, family/friend referrals, and internet searches. Dr. Traversa stated on the 4Q22 call that the company believes patients enrolled from these sources were not adequately vetted with medical or pharmacy records. We believe patients from these questionable sources are inherently more likely to be “doctor-shopping” or otherwise not suffering from true, properly diagnosed MDD, both of which would be more likely to have a placebo response.
Additionally, both RELIANCE-I and RELIANCE-III began enrolling during the COVID-19 pandemic in 2020 and 2021 (before the vaccines were introduced), which dramatically increased rates of situational depression and acute stress, which are also far more likely than true MDD to resolve spontaneously. The company has also stated that internal analyses of RELIANCE-I indicate a significant difference in responses for patients enrolled during versus after COVID lockdowns had ended.
Lastly, consultations with clinical trial design experts concluded that the RELIANCE-I/III study protocols included too many patient assessments and resulted in overly long site visit times, both contributing to expectation bias and increasing placebo responses.
Takeaway
We believe Relmada was overconfident following the Phase 2 results and prioritized enrolling the RELIANCE trials as expeditiously as possible, without due regard for the quality of the trial and accurate diagnosis of the patients. We believe that a suboptimal trial design, lax enrollment criteria and oversight, a concurrent pandemic, and the inclusion of two highly anomalous clinical sites led to suspect data that rendered REL-1017’s efficacy signal undetectable.
Moving Forward: RELIANCE-II and RELIGHT
RELIANCE-II, the second adjunct MDD study (initiated in April 2021), had enrolled only ~80-100 of its intended 300 patients in late 2022 when the failed trials were announced.
Based on the learnings from RELIANCE-I/III, Relmada made several key protocol amendments and process changes to the ongoing RELIANCE-II trial, including:
Exclusion of clinical sites that management deemed to have inferior practices and execution, especially those that reported highly unusual results in RELIANCE-I/III.
Close oversight of clinical sites, including ensuring that patients are evaluated by the same rater at each visit throughout the trial, monitoring staff turnover, and ensuring timely entry of patient data into the electronic data capture system.
Meeting with clinical sites both in person and virtually to ensure protocol adherence.
Requiring proof of prior MDD diagnosis and treatment, including medical and pharmacy records.
Simplifying the study protocol to dramatically decrease the number/frequency of assessments and time spent at trial sites for patients, increasing ease for patients/HCPs and decreasing expectation bias.
Limiting enrollment to “verified” sources, such as patients already in the site’s database and direct referrals from HCPs.
Limiting the number of patients that can be enrolled at any one trial site (the two high-enrolling trial sites contributed 18.5% of RELIANCE-I’s patients).
CMO Cedric O’Gorman gave an example of the proactive approach Relamada is taking at the Goldman Conference, saying: “The oversight is all about being able to contact the site in real time and say this seems off. Can you explain this? Not waiting until the end of the month or the end of the quarter to kind of then collate all your red flags and address them”.
Management has made it clear that it believes its protocol amendments to RELIANCE-II are sufficient to detect an efficacy signal for REL-1017 if present, even with the first ~100 patients being enrolled under the original, unamended trial protocol. Importantly, RELIANCE-II did not enroll any of its patients from the two high-enrolling clinical sites that affected RELIANCE-I/III. Additionally, the vast majority of RELIANCE-II patients were enrolled after COVID restrictions were lifted.
The company also initiated an additional Phase 3 study of REL-1017 as an adjunct therapy in MDD, named RELIGHT, in August 2023, which will be completely “clean” of any issues related to RELIANCE-I/III.
We believe management has taken meaningful and consequential steps to correct the deficiencies of previous trials, which give REL-1017 a much greater chance of demonstrating its true efficacy signal. With RELIANCE-II, RELIGHT, the open-label safety study (discussed below), in addition to the extensive safety data and post-hoc analyses from the failed Phase 3 trials, Relmada would meet the FDA’s total patient data requirement for MDD and be able to file an NDA in the 1H25.
Open-Label Study and Approvable Efficacy in MDD
Our confidence in REL-1017’s underlying efficacy is further bolstered by the open-label safety data that was reported in September 2023, which showed a 16.8 pt MADRS reduction at 1 month in 204 de novo adjunct MDD patients (not rolled over from other RELIANCE studies), deepening to 22.5 pts at 12 months.
September 2023 Press Release 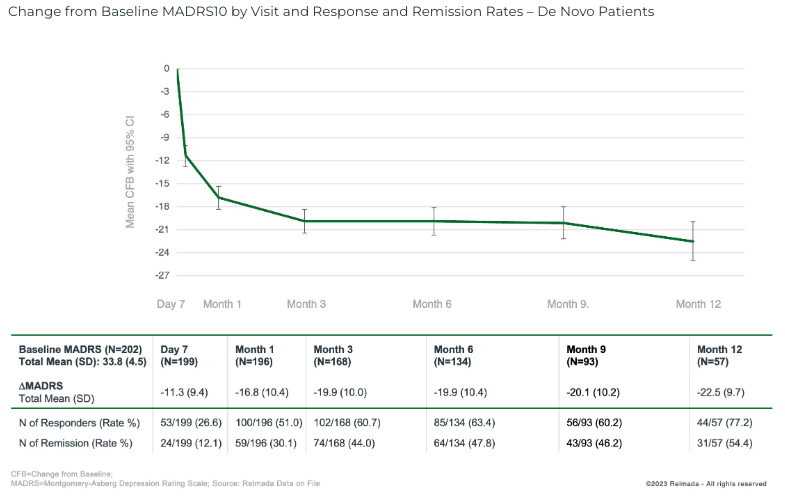
These data further supports REL-1017’s efficacy profile, with REL-1017 reporting robust 16-17 pt MADRS improvements across the open label, Phase 2, and various Phase 3 trial sub-analyses, which span over 400 patients. Even the ~15 pt improvement reported in the RELIANCE-I whole-study data are in-line with many approved antidepressants.
On a placebo-adjusted basis, it has been reported that the average placebo-adjusted improvement on MADRS for approved antidepressants is ~3 pts, with some researchers and retrospective studies claiming just ~2 pts to be sufficient (Thase, et al., 2016).
If the ongoing RELIANCE-II and RELIGHT trials are able to properly control placebo as expected (~8-12 pts of improvement), REL-1017’s 15-17 pts of historical efficacy should be sufficient to deliver a positive result, even with a placebo towards the high end of the typical range.
As an analogue, Axsome’s recently-approved NMDA antagonist Auvelity delivered a 3.9 pt placebo-adjusted improvement at 6 weeks (two weeks longer than REL-1017’s trials) in its pivotal Phase 3 GEMINI trial, which included a similarly-depressed patient population to the RELIANCE studies. The absolute improvement in the Auvelity treatment arm was 15.9 pts at 6 weeks, and ~15.5 at 4 weeks.
Journal of Clinical Psychiatry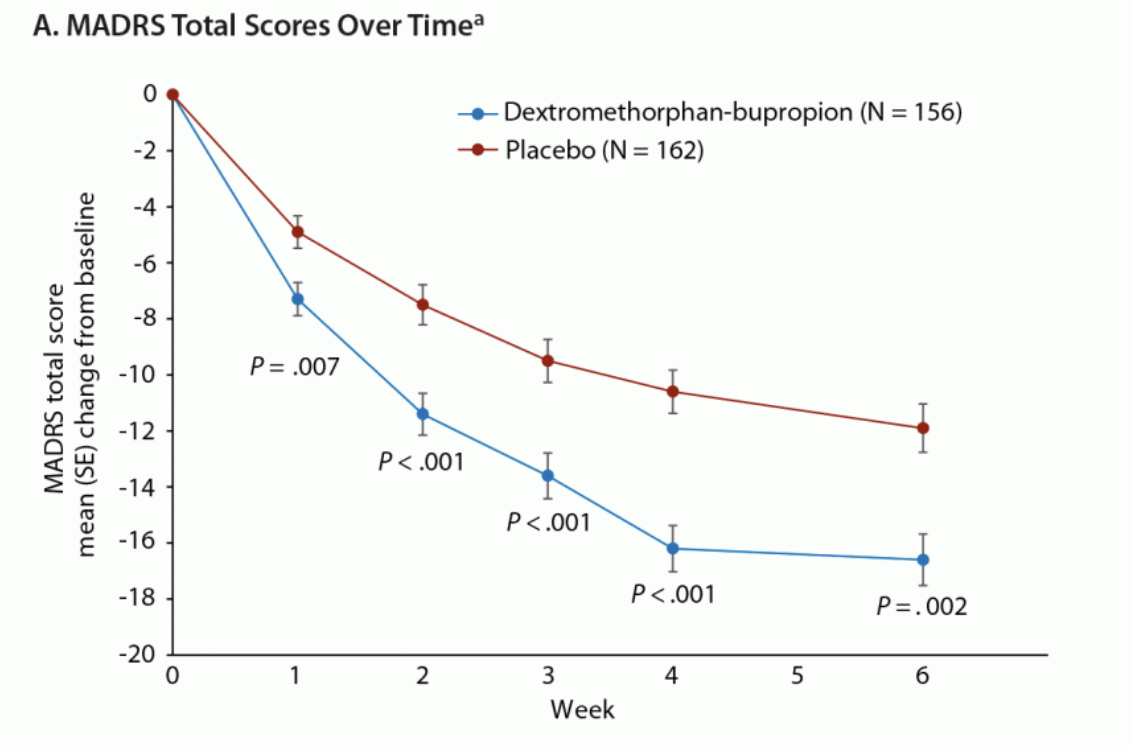
As shown above, the placebo group showed a ~10 pt improvement at 4 weeks, allowing Auvelity’s 15-16 pt improvement to reach deep statistical significance (p<0.001) in a similarly sized trial to RELIANCE-II.
Further, 32% of Auvelity-treated patients achieved clinical remission and 49% achieved clinical response at 4 weeks.
Journal of Clinical Psychiatry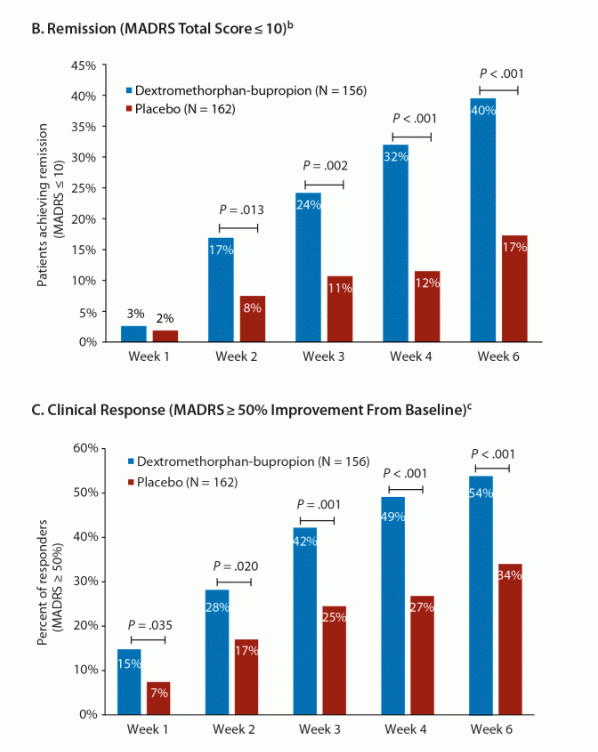
If we assume REL-1017’s Phase 2 data (shown) is closer to what we would expect from Relmada’s ongoing Phase 3 trials, Auvelity and REL-1017 appear to have similar efficacy signals on multiple metrics. (REL-1017’s remission/response data is at Day 14 rather after 7 days of treatment).
Corporate Presentation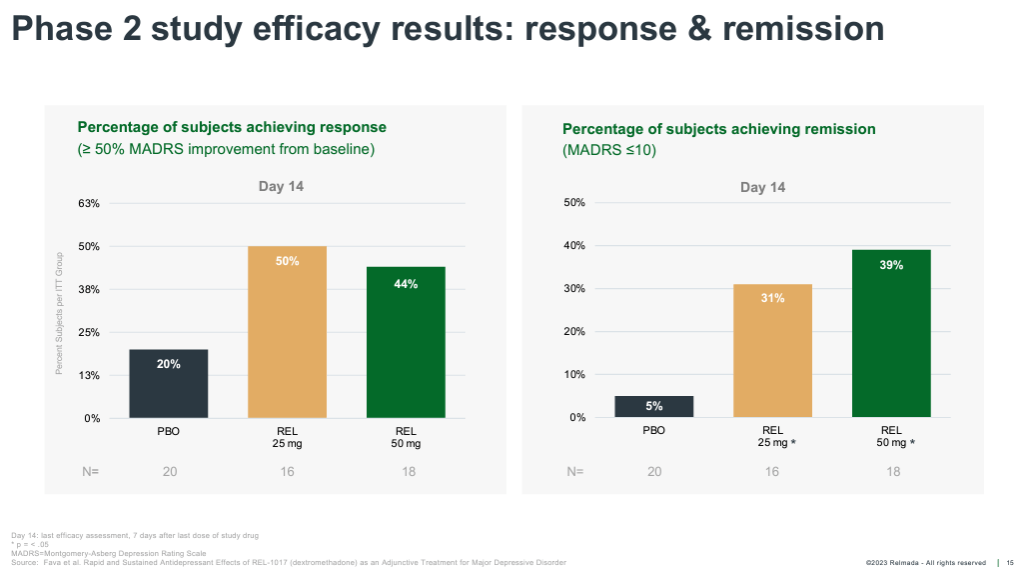
Additionally, Johnson & Johnson’s Spravato, the first NMDA antagonist approved for depression in 2019, reported 4-week placebo-adjusted MADRS improvements ranging from 3.2 to 4.1 pts across its TRANSFORM-I, -II, and -III Phase 3 trials.
Escitalopram (Lexapro), a popular SSRI developed by Lundbeck and approved for MDD in 2002, was found to have delivered a placebo-adjusted MADRS improvement of 3.2 pts in a retrospective study (Thase, et al., 2018).
REL-1017 has consistently demonstrated the ability to deliver 15-17 pts of MADRS improvement, which positions it on-par/favorably among other approved and clinical-stage antidepressants. We believe REL-1017’s efficacy data to date presents a reasonable margin of safety to deliver a positive result in the ongoing, properly placebo-controlled RELIANCE-II and RELIGHT trials.
Adjunct MDD
REL-1017 would be the first NMDA antagonist approved specifically as adjunct therapy in MDD. Spravato is approved as an adjunctive therapy for treatment-resistant depression (TRD) patients, a smaller, later-line diagnosis of patients that have failed two or more antidepressants, and Auvelity is approved as a monotherapy in MDD.
There were an estimated 8.9 million medication-treated MDD patients (i.e. addressable via an adjunct antidepressant) in the US in 2021 (Zhdanava, et al., 2021), about half of the estimated total MDD population in the US, and ~3x the size of the TRD patient population.
Historically, the adjunct MDD market has been occupied by antipsychotics and atypical antipsychotics, including Abilify (Bristol-Myers Squibb), Rexulti (Otsuka/Lundbeck), and Seroquel (Cheplapharm Arzneimittel).
AbbVie’s Vraylar (atypical antipsychotic), first approved for schizophrenia and bipolar disorder in 2015, was approved as an adjunct in MDD in 2022. AbbVie management has commented that the expansion of Vraylar into adjunct MDD has been accelerating and contributed to the $300 million revenue guidance raise that was given on the 2Q23 earnings call.
Relmada believes adjunct MDD is a broader indication than monotherapy, avoiding an issue that a drug like Auvelity could potentially run into, which is the reluctance of HCPs to discontinue a partially-effective antidepressant and switch patients over to a new drug.
In practice, it is likely that Auvelity is prescribed as an adjunctive therapy in some cases, and similarly likely that REL-1017 would eventually be prescribed off-label as a monotherapy in many cases, though we expect Relmada to also pursue a supplemental monotherapy approval.
If REL-1017 is successful in demonstrating its efficacy in the ongoing Phase 3 trials, its tolerability profile could make it an attractive, low-risk option for HCPs in the large population of underserved MDD patients responding inadequately to their antidepressant.
Ketamine and Auvelity Success
Spravato, despite its drawbacks that led to a slow initial launch (e.g. dissociation, in-office dosing), reported $300 million of revenue for the 1H23, making $1+ billion blockbuster status likely in the next couple of years.
Similarly, Axsome’s Auvelity, the only other currently approved antidepressant with a claimed NMDA antagonist mechanism, was launched in October 2022. In the 2Q23, just two full quarters into its launch, Auvelity reported $27.6 million of sales, achieving a $100+ million run rate less than a year after launch. Axsome also announced in August that it would expand its sales force by ~40% to 260 employees in the coming months.
While both Spravato and Auvelity have both had successful launches and provide significant benefits to patients, both present drawbacks/challenges that may not apply to REL-1017.
For Spravato, treatment requires medical supervision and must be administered in a doctor’s office, meaning patients and HCPs have to take significant time out of their day. Dosing is twice per week at the initiation of treatment, and then either every week or every two weeks thereafter. Spravato also comes with well-documented side effects such as dissociation and hallucinations which require patients to have a driver to bring them to and from appointments. These drawbacks may not be as significant in a TRD patient population that is often desperate for relief, but nonetheless require significant time and resources.
Auvelity brings a different, less serious set of potential limiting factors. Firstly, it is dosed twice per day, a potential nuisance for patients vs. a once-per-day option (like REL-1017). Additionally, it has been raised that some HCPs may be skeptical of Auvelity’s components as a combination drug (dextromethorphan and bupropion). It was thought that HCPs may opt to prescribe the two ingredients separately to save patients money, though the prevalence of this practice has not been confirmed as of yet. Additionally, some HCPs may be wary of dextromethorphan’s well-known status as the antitussive ingredient in cough syrup with dissociative effects and abuse potential at higher doses. It is worth noting that esmethadone could face a similar hurdle in needing to educate prescribers on esmethadone’s distinction from methadone and lack of abuse potential.
While each of the three NMDA antagonists has its own strengths and weaknesses, we don’t expect REL-1017’s primary competition to be Spravato or Auvelity. Instead, we expect the NMDA antagonist class as a whole to continue increasing its penetration in the MDD market, taking share from traditional SSRIs, antipsychotics, and other antidepressants. Assuming positive Phase 3 results, we believe REL-1017’s simple, safe, and effective clinical profile could lead to a similarly successful launch to that of Auvelity.
Valuation
With renewed study protocols in place, we expect positive results for RELIANCE-II and RELIGHT in mid-2024 and year-end 2024, respectively, and expect these results to restore REL-1017’s perception in the market as a differentiated novel antidepressant.
As such, we think Relmada could return to a similar valuation to what it held following the promising Phase 2 results, which fluctuated between $500 million and $700 million of market capitalization from late 2019 through 2021. An anticipatory run up to the Phase 3 data readout in 2H22 saw Relmada reach a peak $1.1 billion of market cap.
For context, Sage Therapeutics, whose NDA for its lead asset in MDD, zuranolone, was rejected by the FDA in August 2023, traded at a $2+ billion valuation in 2022-1H23 after initiating its rolling NDA submission in mid-2022. Following the rejection, Sage now trades at ~$1.2 billion, owing to its early- to mid-stage pipeline and strategic partnership with Biogen.
Axsome traded at $1-2 billion of market cap in late 2021 and the 1H22 as AXS-05 (now Auvelity) awaited an FDA approval decision. It had traded as high as ~$3 billion in 2020 after reporting its positive Phase 3 results in December 2019, but experienced multiple delays and requests for manufacturing-related improvements from the FDA which damaged investor sentiment. Axsome traded up to ~$3 billion on Auvelity’s approval in August 2022 and currently trades at ~$3.2 billion. We note that Axsome’s valuation is also bolstered by a mid-to-late stage multi-asset pipeline.
Relmada’s current setup reminds us a bit of Axsome in late-2021/early-2022 as a neuropsych biotech which has lost the confidence of investors following a series of adverse events. While Axsome’s concerns were not related to clinical trial data and were inherently less risky, an analysis of REL-1017’s data and management’s amended trial protocols gives us confidence in Relmada’s ability to deliver positive Phase 3 results, restore investor confidence, and ultimately earn FDA approval.
We think Relmada’s stock will likely see a lift from the $3-4/share range it has occupied for the majority of 2023 as we approach the RELIANCE-II data readout in mid-2024. We believe a definitively positive result for RELIANCE-II would boost the stock at least back to the $500 million market cap level, which would correspond to $16.61/share (+389% from 10/10/23 close). If RELIGHT were to also read out positively towards YE24, we believe Relmada could trade back up towards its prior peak ~$1 billion valuation in 2025 as investor confidence in REL-1017 is restored, which would correspond to ~$33/share (~900% upside).
Risks
Amidst the broader market decline that began in August, the biotech sector has fared especially poorly, with the XBI down over 10% just since the start of September, with even worse performance among smaller names.
Biotechs are highly risky, long duration investments are higher sensitive to interest rates and weakening economic conditions than other sectors. Relmada’s status as a micro-cap biotech is especially risky, though it is supported to some degree by its cash balance which is sufficient to get through two key data readouts, allowing time for market conditions and company-specific sentiment to improve before needing to raise additional capital.
Some other key risks to consider with Relmada:
Clinical trial risk: Even a 15-17 pt improvement in the treatment group in either RELIANCE-II or RELIGHT may still be insufficient to reach statistical significance if the placebo group is not adequately controlled.
Psychiatric clinical trials are notoriously prone to placebo responses and data variance.
Relmada has no meaningful clinical pipeline beyond REL-1017, meaning a negative readout for either RELIANCE-II or RELIGHT would likely result in a significantly lower share price.
Regulators, HCPs, or patients may perceive REL-1017 as an opioid due to its chemical relation to methadone.
It is still unclear exactly what factors contributed most to the high placebo responses in RELIANCE-I and -III.
Lack of a dose-dependent response in the Phase 2 data.
Conclusion
Despite the topline failures of RELIANCE-I and RELIANCE-III, we believe the data suggest that REL-1017’s underlying clinical profile is compelling and that trial failures are attributable to a number of trial design and oversight shortfalls. The Phase 2 and open-label trials, as well as various sub-analyses of the Phase 3 trials, consistently show MADRS improvements of 15-17 pts for REL-1017, on par with the effect sizes of recently approved antidepressants like Axsome’s Auvelity. Assuming that the placebo response is successfully controlled by the amendments enacted by Relamada, we believe REL-1017’s demonstrated efficacy profile is likely to deliver positive results in the ongoing RELIANCE-II and RELIGHT trials. If our thesis plays out, we would expect the market to recognize REL-1017 as an intriguing, novel, approval-stage antidepressant, resulting in significant (~10x) upside potential over the next 12-24 months.
Editor’s Note: This article covers one or more microcap stocks. Please be aware of the risks associated with these stocks.




")






")

")

{kind=link}